近期,我校生物工程学院赵群教授和浙江工业大学杨云芳教授、赫罗纳大学Marc Garcia-Borràs教授、约翰霍普金斯大学Xiongyi Huang教授在金属酶催化新功能拓展方面取得重要进展,研究成果“Engineering non-haem iron enzymes for enantioselective C(sp3)–F bond formation via radical fluorine transfer”正式发表于《Nature Synthesis》杂志。

将氟原子或含氟片段引入药物分子,能够有效地改变药物分子的pKa、脂溶性、代谢稳定性以及渗透性,影响药物分子的吸收、分布和代谢。因此,向药物分子中引入“氟”已经成为药物开发中非常重要的一部分,逐步成为药物筛选的一种常用手段。2021年美国FDA批准的小分子药物中,30%都是有机氟化合物。目前,超过25%的药物都是有机氟化合物。制药工业对含氟化合物的高需求极大地推动了有机氟化学的发展,越来越多的化学合成方法被应用于制备含氟分子,药物开发中的含氟分子数量剧增。
与之相反,自然界中含氟分子的数量却十分有限。到目前为止,人们仅发现约30种天然有机氟化合物,但是同为卤素家族的天然有机氯化合物和有机溴化合物有近2000种已被发现。导致自然界中有机氟化合物稀缺的一个重要原因是酶促碳–氟键形成机制的单一性,目前唯一已知能够在生物合成途径中催化C–F键形成的酶是氟化酶(Fluorinase),它能够催化氟负离子亲核进攻S-腺苷甲硫氨酸(SAM)产生5'-氟脱氧腺苷(5'-FDA)。但是,自然界进化产生的氯化酶和溴化酶可以利用多种反应机制来构建碳–卤键,如:经历次氯酸根中间体的亲电卤代机制、经历铁氧中间体的自由基卤代机制以及亲核卤代机制(图1)。
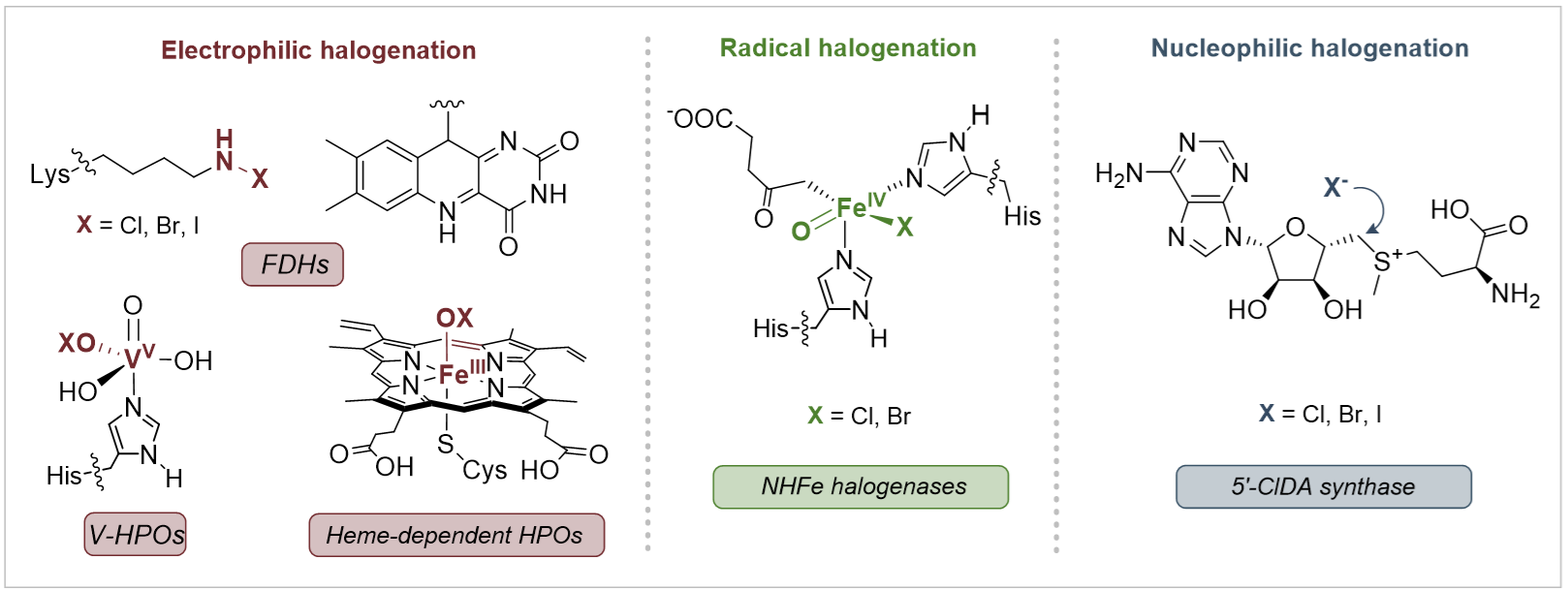
图1:卤化酶催化机制
目前,通过酶促反应合成有机氟化合物的研究,主要集中在拓展氟化酶的底物范围以及将含氟砌块引入代谢途径这两大策略,尽管已经取得了一定进展,但是酶催化C–F键形成的发展仍显不足。如何建立新的生物催化机制来构建C–F键仍然是酶催化领域中颇具挑战性的科学难题。鉴于此,研究团队近日共同报道了基于新催化机制的酶促不对称氟化反应,首次将自由基氟原子转移机制引入到单一金属酶催化体系,实现了高立体选择性的氟转移过程(图2)。
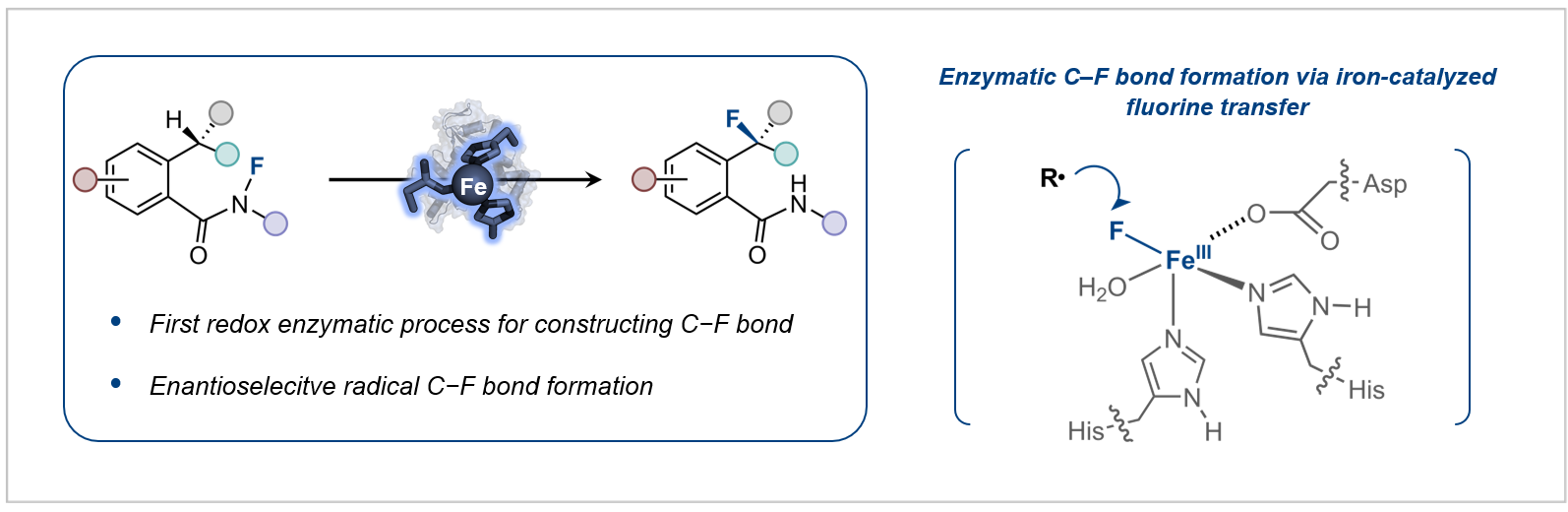
图2:酶催化不对称氟转移反应
首先,作者以N-氟酰胺底物1NF作为模型底物,在全细胞条件下测试了40多种不同的非血红素铁酶,其中包括了前期开发叠氮化反应所积累SavHppD突变体库。通过测试发现了几种能够催化不对称氟转移反应的酶,最好的结果来自绿色链霉菌(Streptomyces viridochromogenes)的(S)-2-羟基丙磷酸酯环氧化酶(SvHppE),它以总转化数TTN = 5、产率3.8%以及e.r = 74:26的对映体比例生成了目标氟化产物。在不含SvHppE的空白对照实验中,仅能观察到痕量的氟化产物。此外,与催化中心铁配位的两个组氨酸被突变为丙氨酸,反应的产率和对映选择性显著降低,上述结果表明该转化过程是由金属铁中心催化完成。
在确认了初始活性后,作者通过定向进化逐步提高了SvHppE的催化性能。鉴于天然底物S-HPP与模型底物N-氟酰胺1NF之间存在较大的结构差异,作者首先构建了SvHppE的同源模型,并对“酶–底物”进行了分子动力学(MD)模拟。再结合隧道分析,作者确定了一些关键的氨基酸残基位点,包括Y101、Y102、A131、N134和F181,这些残基很可能会影响底物1NF在SvHppE中的结合和定位。与中心铁配位的临近氨基酸残基E141同上述五个氨基酸位点,一并作为点饱和突变(SSM)的对象。对E141进行点饱和突变和筛选后发现了E141D优势突变体,其催化活性比野生型明显上升:产率7.1%和对映体比例e.r = 80:20。以E141D突变体作为母体,在其他五个位点上进行了后续点饱和突变和筛选,得到了一个三重突变体SvHppE_N134W_E141D_Y102C(以下简称:SvHppE-Fluor),能够以TTN = 50、产率30%和对映比96.5:3.5的催化效率得到目标氟化产物(图3)。
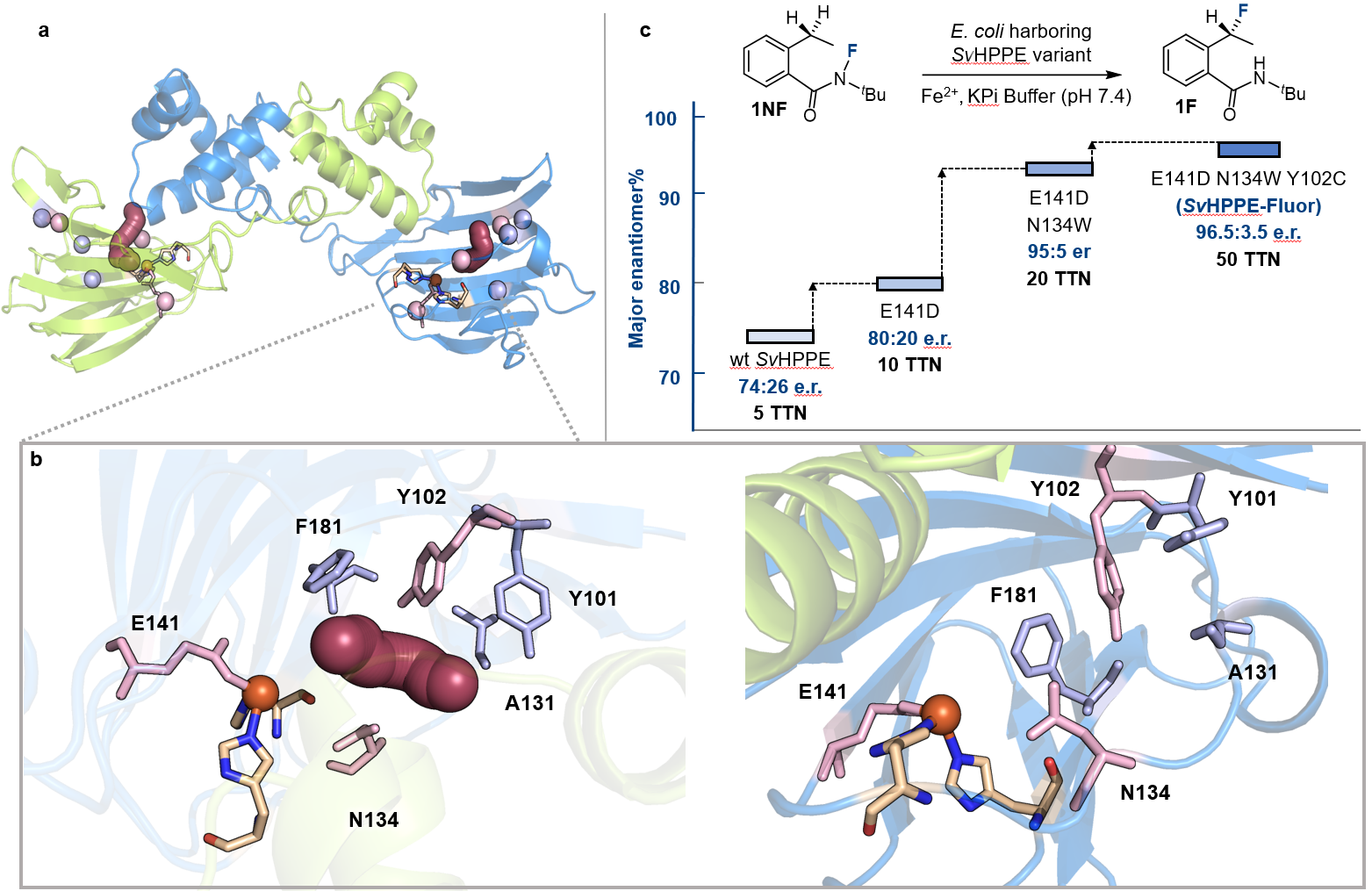
图3:酶定向进化对反应进行优化
接下来,作者对该酶促反应的底物适用范围和局限进行了系统的研究。结果显示,SvHppE-Fluor可以接受多种N-氟酰胺衍生物作为底物并将其转化为相应的氟化产物,该反应对底物芳环上的取代基变化以及烷基取代基链长的变化均表现出了一定的耐受性。当底物芳环对位取代基变化时,立体选择性几乎保持不变;而引入邻位取代基时,立体选择性大幅下降。当底物的烷基取代基链长增加时,反应的转化数会降低,但对映选择性仍然保持在95:5以上(图4)。

图4:酶促反应的底物范围
作者对SvHppE-Fluor和野生型SvHppE进行了分子动力学(MD)模拟(图5),比较了它们的活性位点结构及其与底物1NF的结合情况。MD结果显示,E141D突变显著改变了铁中心的配位环境。通过将谷氨酸E突变为链长略短的天冬氨酸D,中心铁原子将更靠近含有H137、H179和E141D三联体的β桶内。N134W突变对SvHppE活性位点环境的改变更大,在野生型SvHppE中,N134位于活性位点内部,并参与和天然底物S-HPP之间的氢键相互作用,此时的loop1处于开放状态(红色)。而在SvHppE-Fluor中,N134残基变为色氨酸,改变了loop1的构象,使之处于闭合状态(蓝色),此状态有助于维持底物1NF采取有利于反应的姿势。此时底物为顺式构象,羰基氧和氟原子都指向铁中心,这种构象通过铁结合的水分子与羰基之间的氢键相互作用而进一步强化,DFT计算证实了这种配位模式具有能量上的优势。Y102C突变则进一步扩展了活性位点的腔内空间,这一改变使得Y101侧链能够与Y104相互作用,与L143和L192残基一起形成一个疏水口袋,容纳底物1NF的乙基侧链,有利于N–F键活化后的高选择性1,5-氢原子转移过程快速而顺利的进行。

图5:分子动力学模拟(MD)
本文研究团队建立了一种能够介导不对称氟原子转移反应的酶催化体系。这项工作证明非血红素铁酶中的铁–氟中间体能够与碳自由基反应生成碳–氟键,这也预示着其它的非血红素铁酶有望参与自由基型的氟转移反应,能够用于开发非天然途径的氟化反应。该工作为非血红素铁酶的进一步工程化改造提供了借鉴,也为酶促氟化反应提供了新的思路。

























